Cell Metabolism | Xu Daichao’s Team Identifies a Methionine Metabolism Sensor that Regulates Cell Death
Date:2025-06-26
A fundamental question in metabolism is how cells sense nutrients or surrogate metabolites to maintain cellular homeostasis. The capacity of cells to sense and respond to fluctuations in nutrient levels is essential for survival. Various nutrient sensors, which respond to specific nutrient components, have been identified in metabolic pathways. These sensors trigger signaling cascades that adjust cellular processes for metabolic homeostasis. While numerous studies have focused on identifying nutrient sensors in metabolic pathways, the mechanisms connecting nutrient sensing to cell death pathways, such as apoptosis and necroptosis, remain poorly characterized.
Methionine is an essential amino acid for humans. S-adenosylmethionine (SAM), a key metabolite of methionine, is required for normal cellular function and survival. As the primary methyl donor in cells, SAM transfers methyl groups to various substrates, participating in numerous anabolic and catabolic reactions, particularly in one-carbon metabolism. Additionally, SAM plays a vital role in preventing cell death and exhibiting anti-inflammatory properties, though the underlying mechanisms remain unclear.
On June 25, 2025, Xu Daichao’s team at the Interdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, published anarticlein Cell Metabolism titled "RIPK1 senses S-adenosylmethionine scarcity to drive cell death and inflammation". Thisstudy reveals a previously unrecognized role of receptor-interacting protein kinase 1 (RIPK1) as a nutrient sensor, and elucidates the molecular mechanism by which RIPK1 detects methionine and SAM levels to regulate metabolic changes that determine cell fate.

RIPK1 is a key regulator of cell death and inflammation, playing a significant role in various inflammatory and degenerative diseases. In this study, the team screened multiple amino acid deprivation conditions and found that only methionine deprivation specifically induced RIPK1 kinase-dependent apoptosis and inflammation, which could be reversed by SAM supplementation. A mouse model fed a low-methionine diet further validated these findings: wild-type mice exhibited mild inflammation and liver injury, while RIPK1 kinase-dead mutants (Ripk1-D138N) completely alleviated these pathologies.
Moreover, the researchers discovered that RIPK1 directly senses changes in SAM levels. When methionine metabolism or one-carbon metabolism is disrupted, leading to reduced SAM levels, RIPK1 is activated to initiate apoptosis and inflammation. Specifically, SAM, as a methyl donor, facilitates symmetric dimethylation of RIPK1 at arginine R606, catalyzed by protein arginine methyltransferase 5 (PRMT5), thereby inhibiting RIPK1 activation.
RIPK1 consists of an N-terminal kinase domain, an intermediate domain, and a C-terminal death domain (DD). The R606 residue is located in the death domain and is highly evolutionarily conserved. Previous studies suggested that RIPK1 undergoes autoactivation through oligomerization of the death domain, but the mechanism remained unclear. Using cryo-electron microscopy, the team resolved the oligomerization interface structure of RIPK1-DD and found that the R606 residue directly participates in RIPK1 oligomerization, which is critical for its activation. Thus, under SAM-sufficient conditions, methylation of R606 suppresses RIPK1 oligomerization and activation. Conversely, when methionine or SAM is scarce, unmethylated RIPK1 readily oligomerizes and activates, triggering cell death and inflammation (Figure 1). In Ripk1-R606K mutant mice, the inflammatory and liver injury phenotypes induced by a low-methionine diet were significantly suppressed. Conversely, hepatocyte-specific PRMT5 knockout led to aberrant RIPK1 activation and spontaneous hepatitis, liver injury, and hepatocellular carcinoma progression, all of which were completely reversed by the Ripk1-D138N mutation.
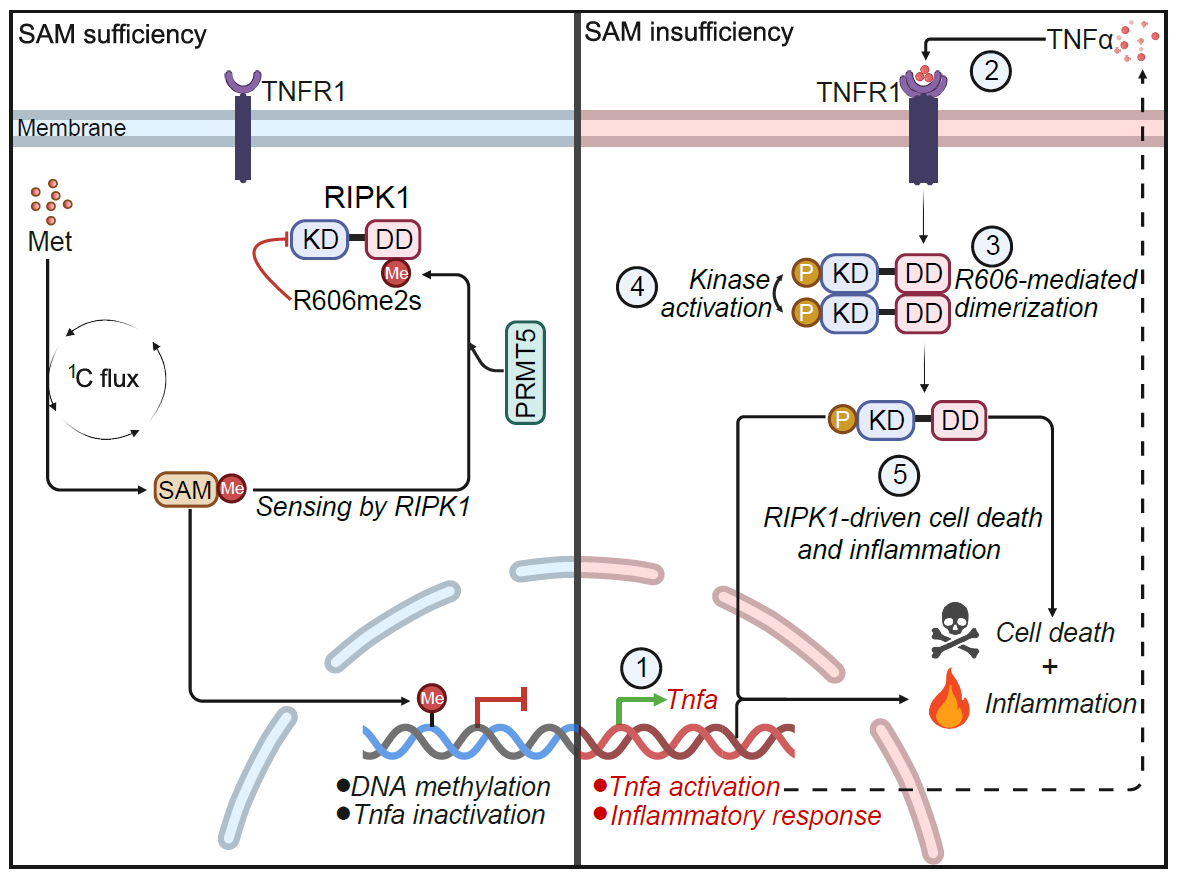
Figure 1. Mechanism model of RIPK1 sensing methionine and SAM levels to regulate cell fate and inflammation.
In summary, this study identifies RIPK1 as a novel sensor for SAM abundance, capable of responding to methionine and SAM metabolic changes to control cell fate decisions. SAM deficiency contributes to the development of diseases related to cell death and inflammation. Given that RIPK1 inhibitors have shown promise in clinical trials for various inflammatory and degenerative diseases, this research suggests RIPK1 as a potential therapeutic target for human diseases associated with SAM dysregulation.
The corresponding author of this study is Researcher Xu Daichao from the Interdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences. Professor Gu Jinyang from Tongji Medical College, Huazhong University of Science and Technology, serves as co-corresponding author. The first author is Chen Zezhao, a Ph.D. student at the Interdisciplinary Research Center on Biology and Chemistry, and Professor Gu Xiaosong from the Second Affiliated Hospital of Soochow University is listed as co-first author. Researchers Zhang Yixiao, Zhu Zhengjiang, and Associate ResearcherLiu Jianping from the Interdisciplinary Research Center on Biology and Chemistry also provided valuable contributions to this work.
Link to the original article: https://doi.org/10.1016/.cmet.2025.05.014
附件下载: