Advanced Science | Zhang Yaoyang’s Team Develops DeepSecMS for In-Depth Selenoproteome Profiling
Date:2025-07-29
Selenoproteins are a special class of proteins that contain selenocysteine (Sec, U), often referred to as the 21st amino acid. Sec is a structural analogue of cysteine (Cys, C) but possesses unique chemical properties that enable selenoproteins to play essential roles in maintaining cellular redox homeostasis and regulating key physiological processes. They are closely associated with various diseases, including neurodegenerative disorders, cancer, cardiovascular diseases, and diabetes. In-depth characterization of selenoproteins is therefore critical for elucidating the molecular mechanisms underlying these diseases. However, due to the rarity of Sec and the analytical challenges in detecting Sec-containing peptides, only 25 human selenoproteins have been identified to date, making the creation of a complete selenoprotein atlas and the discovery of novel selenoproteins a major challenge.
Previously, the research group led by Prof. Yaoyang Zhang at the Interdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, developed a Sec-specific mass spectrometry method (SecMS) and a SECIS-independent selenoprotein database (SIS) (PMID: 30174312). Using these tools, they created the first tissue-specific selenoprotein atlas in mice and discovered multiple novel selenoproteins, providing important theoretical and data resources for systematic selenoproteomics research. Recently, the Zhang group published a study in Advanced Science titled “DeepSecMS Advances DIA-Based Selenoproteome Profiling Through Cys-to-Sec Proxy Training”. In this work, the researchers developed the DeepSecMS method based on deep learning and large-scale proxy data training, enabling in-depth profiling of the mammalian selenoproteome.

Data-independent acquisition (DIA) mass spectrometry has recently gained attention for its ability to provide comprehensive data collection, along with accurate and reproducible quantification. Conventional DIA analyses often require spectral libraries generated through data-dependent acquisition (DDA) experiments, which can be time-consuming and incomplete. More recently, advances in deep learning have enabled the prediction of in silico spectral libraries, providing a DDA-free, accurate, and comprehensive alternative for both regular and modified peptides. The prediction-based strategy is extremely appealing, especially for identifying novel selenopeptides that have never been captured through DDA analysis. However, deep learning-based spectral prediction cannot be easily applied to selenopeptides due to the limited number of spectra for known selenopeptides, which severely hinders the accuracy of model training.
To address this limitation, the researchers innovatively developed the DeepSecMS workflow (Figure 1). Given the rarity of Sec and its chemical similarity to Cys, they utilized a proxy training strategy using a large dataset of Cys-containing peptides to generate a large-scale theoretical library of Sec-containing peptides. They demonstrate that DeepSecMS enables the accurate prediction of critical features of Sec-containing peptides, including MS2, retention time (RT), and ion mobility (IM). By integrating DeepSecMS with DIA methods, the identification of known selenoproteins was significantly enhanced across diverse cell types and tissues. More importantly, it facilitates the identification of numerous highly scored, potential novel selenopeptides. These findings highlight the powerful potential of DeepSecMS in advancing selenoprotein research. (Figure 2)
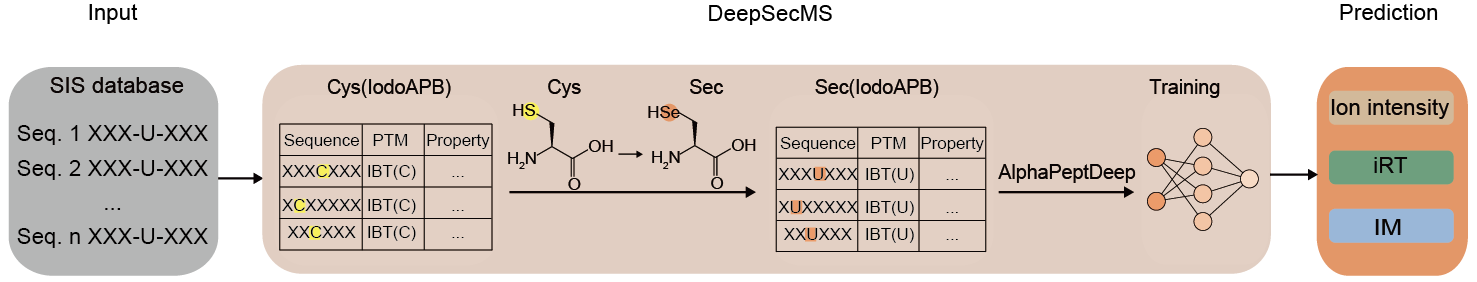
Figure 1. DeepSecMS workflow.
In summary, the newly developed DeepSecMS method represents a powerful tool for selenoprotein identification and holds great potential for discovering novel selenoproteins. This innovative approach signifies a substantial advancement in selenoproteomics, offering new tools into selenoprotein exploration and its potential roles in human health and disease. Moreover, this proxy training strategy may be extended to the analysis of other rare post-translational modifications, providing a scalable technical framework to advance trace proteomics research.
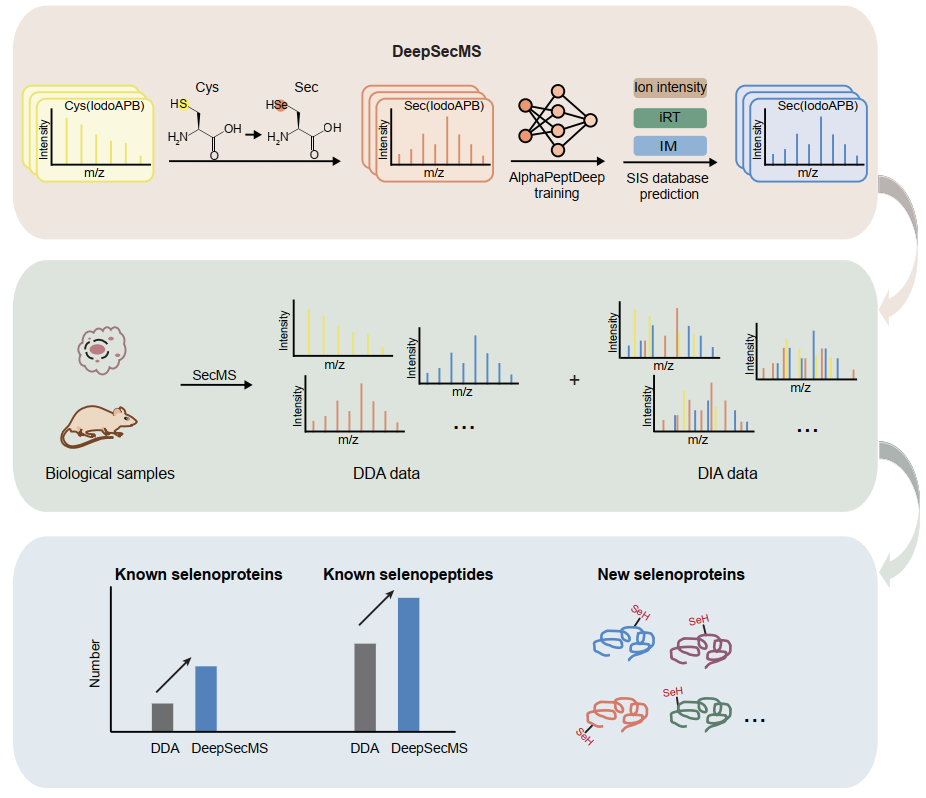
Figure 2. In-depth profiling of the mammalian selenoproteome using DeepSecMS.
Prof. Yaoyang Zhang from the Interdisciplinary Research Center on Biology and Chemistry, Chinese Academy of Sciences, is the corresponding author of this work, and Dr. Chenfang Si is the first author. Prof. Wen-Feng Zeng from Westlake University provided important technical support, and Prof. Liang Qiao from Fudan University offered valuable assistance. This work was supported by the National Natural Science Foundation of China, the Chinese Academy of Sciences, and the Shanghai Municipal Science and Technology Commission.
Original article link: http://doi.org/10.1002/advs.202504109
附件下载: