The Cong Liu team and collaborators develop a new biomarker-based method for synucleinopathy subtyping
Date:2026-05-27
Neurodegenerative diseases (NDs) are a group of progressive neurological disorders closely associated with aging. A common pathological hallmark of NDs is the misfolding of specific proteins and the formation of abnormal amyloid aggregates1,2. Pathological aggregates can propagate and amplify between cells, thereby accelerating disease progression. Synucleinopathies represent a class of NDs characterized by the abnormal aggregation of α-synuclein (α-syn), among which Parkinson’s disease (PD) and multiple system atrophy (MSA) are the most representative disorders3,4. Currently, the diagnosis of synucleinopathies relies primarily on clinical assessment. However, the clinical symptoms of different subtypes overlap substantially, and clinical manifestations do not directly reflect differences in underlying molecular pathology. Therefore, new clinical diagnostic methods based on key pathological biomarkers are urgently needed for early diagnosis and precise disease subtyping.
In recent years, Alzheimer’s disease (AD) research has developed a robust clinical diagnostic paradigm centered on the two key pathological biomarkers, amyloid β (Aβ) and Tau, integrating fluid biomarker testing with brain PET imaging. This paradigm has markedly improved the specificity and accuracy of disease diagnosis, biomarker-based classification and staging, and has directly accelerated the development and clinical approval of disease-modifying therapies targeting critical AD pathologies5,6. By contrast, the biomarker-based diagnostic framework for synucleinopathies, including PD and MSA, remains substantially less developed than that for AD. Clinically applicable fluid biomarker assays and brain PET imaging methods are still lacking, which has substantially hindered the development of disease-modifying therapies for synucleinopathies.
On May 26, 2026, Prof. Cong Liu’s team at the Interdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, in collaboration with Prof. Dan Li’s team at the Bio-X Institutes, Shanghai Jiao Tong University, and Prof. Jian Wang’s team at Huashan Hospital, Fudan University, published a research article in Cell entitled “TPPP/p25 amyloid seeding activity as a specific biomarker for multiple system atrophy”.

PD and MSA both belong to the spectrum of synucleinopathies, but they differ markedly in their cellular pathology. PD is mainly characterized by the accumulation of α-syn aggregates in neurons, forming Lewy bodies and Lewy neurites (LBs/LNs). In contrast, MSA is typically defined by α-syn aggregation in oligodendrocytes, forming glial cytoplasmic inclusions (GCIs). Based on this distinction in cellular pathology, the research team focused on TPPP/p25, an oligodendrocyte-enriched protein, and used an integrated set of biochemical, biophysical, cryo-EM and clinical CSF sample testing to investigate its amyloid aggregation mechanism and establish the TPPP/p25-SAA.
The main findings are as follows: (1) Under physiological conditions, full-length TPPP/p25 possesses an intrinsic self-protective mechanism and does not form amyloid fibrils spontaneously; (2) The CORE domain is the key region that mediates TPPP/p25 amyloid aggregation and undergoes substantial conformational rearrangement during fibril formation. (3) The disease-associated A119V mutation enhances the aggregation propensity of TPPP/p25, suggesting that the self-protective conformation of TPPP/p25 may be disrupted by disease-related factors, thereby promoting amyloid aggregation. (4) Guided by mechanistic and structural information, the researchers rationally designed and screened a CORE-domain-derived miniCORE construct as an efficient amplification substrate, establishing the TPPP/p25-SAA detection method; (5) TPPP/p25-SAA specifically detects pathological TPPP/p25 seeds in CSF, shows no significant response to other amyloid seeds such as Aβ, Tau and α-syn, and effectively distinguishes MSA from PD, dementia with Lewy bodies (DLB) and healthy controls (HC). These findings demonstrate its potential clinical application as an MSA-specific CSF biomarker and a molecular subtyping tool for synucleinopathies.
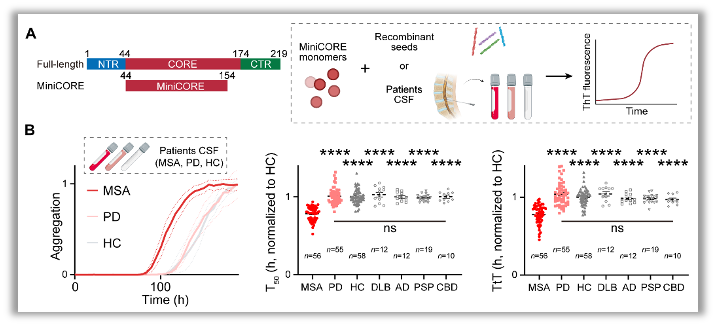
Figure 1. Schematic of TPPP/p25-SAA and analysis of clinical CSF samples from different disease cohorts.
A. Domain organization of miniCORE (left) and the cartoon workflow of TPPP/p25-SAA (right).
B. Representative aggregation curves of SAA with the addition of MSA (red), PD (pink), and HC (gray) CSF (left). Data are represented as mean ± SEM, n = 3/4 individual samples. T50 (time to 50% maximal fluorescence, middle) and TtT (time to threshold, right) for MSA (n = 56, red), PD (n = 55, pink), HC (n = 58, gray), DLB (n = 12, gray), AD (n = 12, gray), progressive supranuclear palsy (PSP; n = 19, gray), and corticobasal degeneration (CBD; n = 10, gray). Each dot represents an individual biological sample. Data are represented as mean ± SEM. Values are compared using one-way ANOVA followed by Tukey’s post hoc test. ns, no significant differences for comparisons among PD, HC, DLB, AD, PSP, and CBD groups; ****p < 0.0001 versus MSA.
In summary, TPPP/p25 amyloid aggregates with seeding activity in patient CSF show potential as a novel biomarker for synucleinopathies. This study elucidates the molecular mechanism of TPPP/p25 amyloid aggregation, determines its fibril structure, and establishes TPPP/p25-SAA based on these mechanistic and structural insights. TPPP/p25-SAA effectively distinguishes MSA from HC and further differentiates MSA from PD, thereby addressing a key limitation of α-syn-SAA in synucleinopathy subtyping, particularly in the differential diagnosis of MSA and PD7,8.
Notably, in AD, combined assessment of Aβ and Tau biomarkers has improved disease diagnosis, biomarker-based subtyping and staging, and promoted the clinical translation of therapies targeting AD pathologies. Similarly, combining TPPP/p25-SAA with α-syn-SAA may provide a dual-biomarker strategy for synucleinopathies by capturing pathological changes at different molecular levels. This approach could improve diagnostic accuracy, subtyping reliability and patient stratification, and provide a molecular diagnostic tool for MSA-PD differential diagnosis, clinical trial enrollment and disease-modifying therapy development. Further clarification of how pathological TPPP/p25 aggregates are generated, released and propagated in CSF, as well as how TPPP/p25 interacts with α-syn under pathological conditions and influences disease progression, will deepen our understanding of synucleinopathy pathogenesis and provide a new theoretical basis for molecular diagnosis and potential therapeutic intervention.
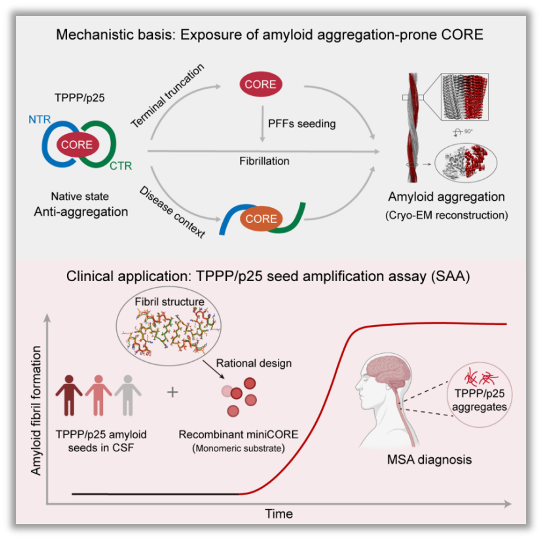
Figure 2. Molecular mechanism of TPPP/p25 self-protective mechanism and the establishment and clinical application of TPPP/p25-SAA
Under physiological conditions, the disordered N-terminal region (NTR) and C-terminal region (CTR) of TPPP/p25 stabilize the aggregation-prone CORE domain through intramolecular interactions, thereby maintaining a self-protective conformation and suppressing amyloid aggregation. Under pathological conditions, this self-protective conformation is disrupted, promoting pathological TPPP/p25 aggregation. Based on the cryo-EM structure of pathological TPPP/p25 fibrils and the molecular mechanism of CORE-mediated aggregation, miniCORE was rationally designed as an SAA amplification substrate, leading to the establishment of the TPPP/p25-SAA detection system. This assay enables the detection of pathological TPPP/p25 amyloid seeds in patient CSF and supports the specific molecular diagnosis of MSA.
This study was jointly supervised by Prof. Dan Li from the Bio-X Institutes, Shanghai Jiao Tong University; Prof. Cong Liu from the Interdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences; and Prof. Jian Wang from Huashan Hospital, Fudan University. The co-first authors are Shuyi Zeng, a 2022 PhD student at the Bio-X Institutes, Shanghai Jiao Tong University; Shenqing Zhang, a 2025 PhD graduate from the Bio-X Institutes; Dr. Shengnan Zhang, an associate research professor in the Cong Liu laboratory; Dr. Yun Fan from Huashan Hospital, Fudan University; Wencheng Xia, a 2025 PhD graduate from the Cong Liu laboratory; and Feiyang Chen, a 2023 PhD student at the Bio-X Institutes. This work was supported by the National Natural Science Foundation of China, the Ministry of Science and Technology of China, the Science and Technology Commission of Shanghai Municipality, the Shanghai Academy of Natural Sciences (SANS), and the Chinese Academy of Sciences.
Original article: https://www.cell.com/cell/fulltext/S0092-8674(26)00517-9
REFERENCES
1. Wilson, D.M., 3rd, Cookson, M.R., Van Den Bosch, L., et al. (2023). Hallmarks of neurodegenerative diseases. Cell 186, 693-714.
2. López-Otín, C., Blasco, M.A., Partridge, L., et al. (2023). Hallmarks of aging: an expanding universe. Cell 186, 243-278.
3. Bloem, B.R., Okun, M.S., and Klein, C. (2021). Parkinson's disease. Lancet 397, 2284-2303.
4. Krismer, F., Fanciulli, A., Meissner, W.G., et al. (2024). Multiple system atrophy: advances in pathophysiology, diagnosis, and treatment. Lancet Neurol 23, 1252-1266.
5. Jack, C.R., Jr., Knopman, D.S., Jagust, W.J., et al. (2013). Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12, 207-216.
6. Therriault, J., Schindler, S.E., Salvadó, G., et al. (2024). Biomarker-based staging of Alzheimer disease: rationale and clinical applications. Nat Rev Neurol 20, 232-244.
7. Fairfoul, G., McGuire, L.I., Pal, S., et al. (2016). Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol 3, 812-818.
8. Grossauer, A., Hemicker, G., Krismer, F., et al. (2023). α-Synuclein seed amplification assays in the diagnosis of synucleinopathies using cerebrospinal fluid-a systematic review and meta-analysis. Mov Disord Clin Pract 10, 737-747.
附件下载: